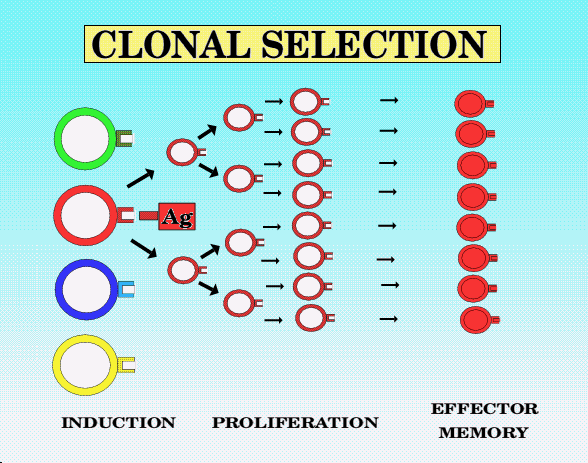
Clonal selection
The whole basis of specificity within the immune system is that clones of lymphocytes are generated with random receptors for antigen. Within this random pool of receptors, some will have high affinity for self molecules, and these clones will normally be deleted at the site of their generation (ie. the thymus for T-lymphocytes). Other clones, by chance, will have high affinity for particular molecules (antigens) on, for example, an infecting flu virus, and this will allow the virus to be recognised by these clones. Recognition and activation then causes clonal expansion, so that a much higher proportion of T-lymphocytes will be available to recognize any further infection with the same flu virus, providing the basis of immunological memory. However, because the receptors are generated randomly, many will have a moderate affinity for both foreign proteins on infectious organisms, but possibly also a moderate affinity for self proteins not present at their site of generation (ie. non thymic antigens, in the case of T-lymphocytes). It is in considering how the immune system can distinguish between self and non-self in the case of these potentially cross-reactive clones that allows us to understand autoimmune diseases and how they might be treated.

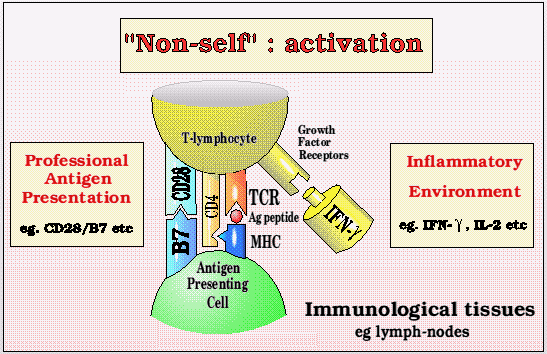
T cell and APC interaction molecules
A number of other adhesion molecules and growth factors are also used to send signals between the T- lymphocyte and the Antigen Presenting Cell, and only if all the signals are correct does the T-lymphocyte become activated and aggressive. An aggressive response results in the multiplication of that clone of T- lymphocytes, which also develop the ability to kill all further infected cells, using a similar recognition process to that shown above.
However, most of our body is not structured like a lymph-node, and if healthy should not have inflammation, and in this situation many of the important molecules for signalling aggression are not expressed. In this situation, although antigen can still be recognized by the T-lymphocyte, the response is not one of aggression, but rather of tolerance.
This non-inflammatory environment can be reinforced by the presence of anti-inflammatory cytokines like IL-4 and IL-10, that can either be produced by healthy tissues, or by a population of T-lymphocytes that are protective and tend to yet further suppress any tendency to (self) aggression. We will return to the concept of suppressor T-lymphocytes below.
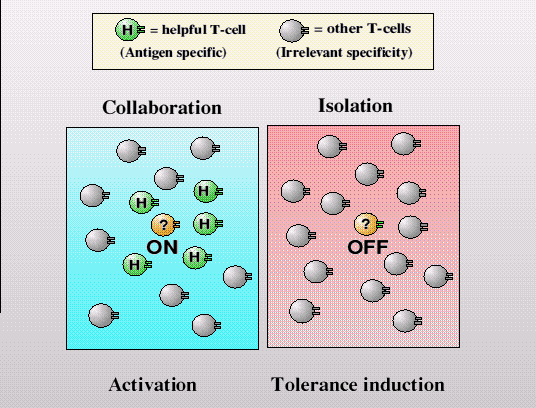
T-lymphocytes are similar in that unless sufficient numbers of them are helping each other become aggressive in the same local environment they instead default to the tolerant, perhaps even protective state. For normal body antigens, the vast majority of T-lymphocytes will already be in a state of self-tolerance, generated during development, and so ensuring that only very few, potentially auto-agggressive T-lymphocytes arise, and like hooligans isolated in a crowd they will be unable to cause much trouble on their own. Infectious organisms, however, will suddenly present a whole range of new foreign antigens to the immune system, able to be recognized by many different T-lymphocytes simultaneously, thereby providing the necessary incitement to aggression.
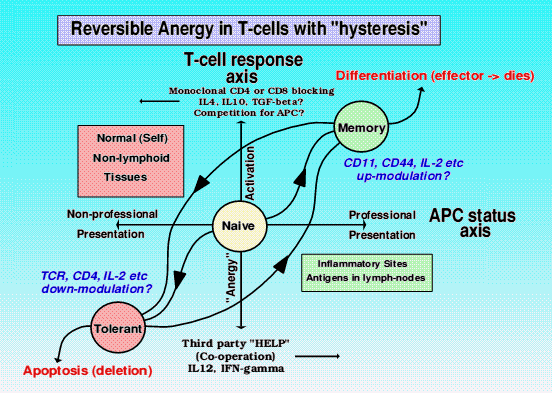
Reversibility of tolerance/activation
The idea of hysteresis, where the degree of response (positive aggression or negative tolerance on the Y axis) resulting from the degree of stimulus (on the X axis) can be used to demonstrate how any state between the two extremes of terminal differentiation and apoptosis can be reversed, but that there is a large resistance to such change. In other words, tolerant T-lymphocytes require a major extra inflammatory stimulus, such as a life threatening viral infection throughout the body, in order to reverse tolerance and hopefullly provide some extra (cross-reactive) immunity in such a dire situation. If, after the infection clears up, these reactivated T-lymphocytes are NOT returned to tolerance, then this is one way in which an autoimmune disease could result. Primed or previously activated T-lymphocytes can conversely be made tolerant if we can efficiently block their reactivation signals. This we can achieve with monoclonal antibodies against these signally molecules (eg CD4 and CD8), is the basis of many of our tolerance model systems in vivo, and holds out some hope to a clinical treatment of autoimmunity and graft rejection.
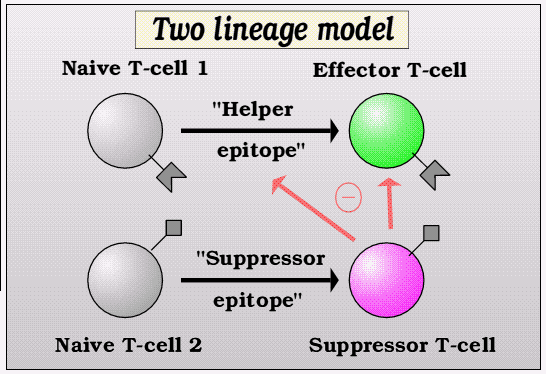
More recently, it has become apparent that suppressor T-lymphocytes may simply represent an alternative path of differentiation than that to effector cells, and may even be identical to those T-lymphocytes that have resulted from a non-aggresive response by becoming tolerant. This alternative differentiation causes the T- lymphocyte to make a different pattern of growth factors or cytokines, that can either directly suppress aggressive T-lymphocytes or may act to make the local environment less inflammatory.
These two alternative pathways of differentiation are somtimes termed Th1 for the aggressive pathway making inflammatory cytokines like interferon-gamma and tumour necrosis factor, and Th2 (or the newly recognized Tr1 subset) for the protective pathway where IL-4, IL-10 and TGF-beta are made to inhibit inflammation.

Infectious tolerance - Th1/Th2 hypothesis
One hypothesis that could explain infectious tolerance is that Th2-type T-lymphocytes make up the tolerant population, and make cytokines like IL-4 that are known to influence naive T- lymphocytes to follow the same pathway of differentiation while at the same time suppressing the generation of aggressive (Th1) responses. Therefore, Th2 cells beget Th2 cells beget more Th2 cells etc.... This is generally known as "Immune Deviation". A more recent idea is that there is a special T cell subset that regulates Th1 responses, termed Tr1 that is induced by IL-10 and acts to down regulate antigen presentation by making cytokines such as TGF-beta. This is perhaps a more attractive hypothesis because we and others have recently shown that Th2 cells are not necessarily protective. For example, both Th1 and Th2 T cell clones directed against the H-Y antigen can reject male skin grafts after transfer in back in vivo.
The hypothesis that it is the balance of Th2 (or Tr1) over Th1 responses that leads to tolerance, and explains its infectious nature is very topical, but it is almost certainly only one part of the mechanism of suppression and immunoregulation. For example, we do not yet know how suppression is able to operate between the two major subsets of T-lymphocytes that express either CD4 or CD8 and only recognize antigen presenting cells or targets that have the appropraite Major Histocompatability Complex molecules (MHC Class II and MHC- Class I, respectively).
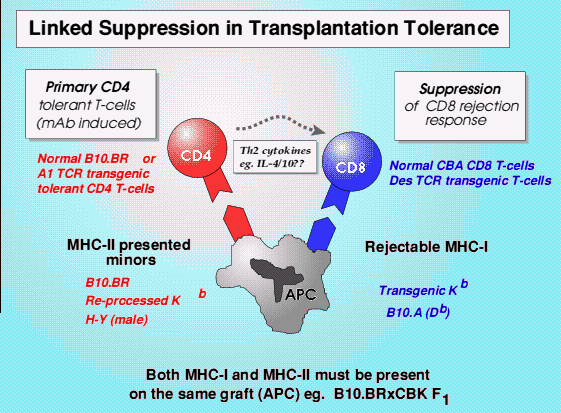
It is now possible to use populations of T-lymphocytes all expressing the same (transgenic) antigen receptor, for example against the male antigen on MHC Class-II cells (H-Y) or a defined MHC Class-I antigen (Kb), and look at the requirements for them to regulate each other. It is this type of experiment that has demonstrated that T-lymphocytes tolerant to one antigen can suppress another set of T-lymphocytes specific for a completely different antigen, if the two antigens are linked on the same antigen presenting cell, presumably bringing them into very close proximity.
Over the last few years it has become clear that there are more functional subsets of helper T-lymphocytes than just the original Th1, Th2 and Tr1. In particular, we now think that in the presence of transforming growth factor beta (TGFbeta), an alternative pathway of differentiation can generate a whole new set of T lymphocytes including regulatory T cells (Tregs), Th17 cells and Th9 cells. The Treg cells, which can be recognised by their expression of the master regulator gene foxp3, are now known to be important in immune tolerance both to self antigens (to control autoimmune disease) and to foreign antigens (to control allergy and maintain transplantation tolerance). Like the Tr1 cells in the infectious tolerance experiments above, Tregs express high levels of CTLA4 and also produce anti-inflammatory cytokines such as TGFbeta and IL-10. We have shown that all these can act to induce enzymes in dendritic cells and other cells within the tissues that consume essential amino acids in a manner similar to IDO catabolising tryptophan. This depletion of essential amino acids is then sensed by both GCN2 (as above) as well as by another molecule called mTOR, and this not only stops the T lymphocytes from proliferating and differentiating into harmful effector T cells, but actually pushes them to express foxp3 and become a new cohort of Tregs. We believe this is an important molecular mechanism which explains how infectious tolerance works. It is also interesting to note that the immunosuppressive drug rapamycin is an mTOR inhibitor, and this may explain why it is one of the few conventional immunosuppressive drugs thought to be tolerance permissive.
In summary, the immune system has developed many mechanisms of distinguishing between self tissues and foreign infections. We are able to exploit and expand some of these natural mechanisms using, for example, monoclonal antibodies that block critical signalling molecules on the T-lymphocytes, such as CD4. This should allow us to develop treatments to re-establish self tolerance in autoimmune diseases, or to generate specific tolerance to foreign organ grafts.
I hope this has been of some use to you, and that I have not managed to put you off immunology for life! If you are confused but still interested why not go back and read the simple version again?
Return to TIG Home Page
Steve Cobbold - updated 29th June 2009.
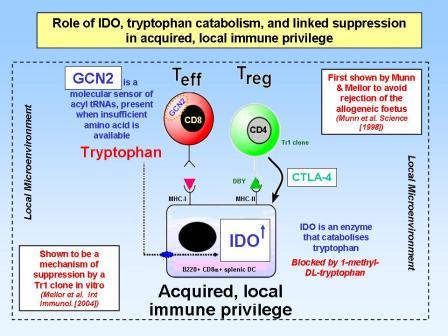
Linked suppression via IDO and tryptophan catabolism
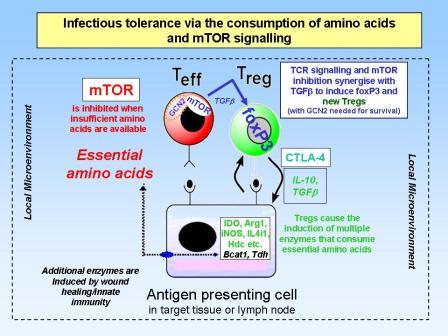
Infectious tolerance via amino acid consumption and inhibition of mTOR signalling
Summary