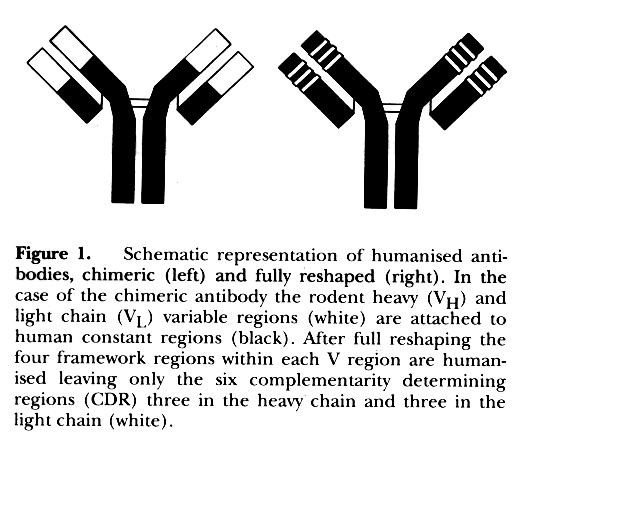
 Figure 1.
Figure 1.
The need to overcome the immunogenicity problem of rodent antibodies in clinical practise has resulted in a plethora of strategies to isolate human antibodies. If human antibodies are to be used, then one would like to understand the basis by which different isotypes interact with host effector systems, and, if possible, improve on nature by engineering in desirable modifications.
The original humanisation strategy described by Winter (1) exploited knowledge of the solved crystal structure with grafting of rodent complementarity determining regions (CDRs) into the defined human frameworks, and judicious mutations in critical framework residues.
In order to humanise successfully many other rat and mouse antibodies we adopted a best-fit strategy whereby we selected the most homologous human framework sequence we could find in the databases, and reshaped to that (2,3,4). The detailed arguments for and against this strategy have been reviewed (101). We have successfully reshaped six antibodies by this route, and failed with only one.
There is still uncertainty about which effector mechanisms determine antibody-mediated cell lysis in vivo. In vitro readouts such as Antibody Dependent Cell-mediated cytotoxicity (ADCC) or complement activation have proven useful in defining differences between isotypes, and in screening of improved antibodies. Because antibodies to the abundant lymphocyte antigen CDw52 (CAMPATH-1) exhibit potent in vitro and in vivo lytic potential (1) they have served as the gold standard by which all modifications or mutations could be compared. Each human IgG has 3 homologous constant-region domains (CH1, CH2 and CH3) and a hinge region between CH1 and CH2 whose size varies between the isotypes. Our laboratory was amongst the first to use matched sets of human antibodies to look for functional differences between the isotypes (5).
By using domain-switch antibodies (6,7 , 8) it can be shown that the differential ability of the isotypes to activate complement depends largely on the carboxy-terminal half of the CH2 domain. Likewise, we have also shown that this half of the CH2 domain is critical to ADCC through the FcRIII receptor. The binding sites for FcR1 and FcRII have both been shown to involve residues in the hinge-link region and the C-terminal half of the CH2 domain. We have also discovered an interesting polymorphism in human ADCC effector function, whereby some individuals can use all IgG isotypes, while others perform poorly with all the isotypes except for IgG1 (6).
It is known that to achieve efficient monoclonal antibody-mediated killing, a high antigen-density is advantageous, presumably by approximating sufficient Ig effector domains together. With the knowledge that the CH2 and hinge regions have an important role in effector functions, a series of multiple-domain antibodies that contain extra CH2 and hinge regions have been created (9) and are being investigated for any changes in effector potency.
None of the in tandem series of CAMPATH-1 antibodies demonstrated better complement lysis than the parent, while the CH1-hinge-CH2-CH2-CH3 form actually lost activity. But an alternative approach providing extra domains in parallel has recently been described by Shopes (10) through creation of dimers by engineering a cysteine into the H-chain of a chimeric Ig at position 444. The new disulphide bonds were able to link pairs of Ig tail to tail to form covalent dimers. These novel antibodies were found to have enhanced lytic ability with heterologous complement. Such a dimeric construct has been created for CAMPATH-1H (9) , and although no more efficient than the monomer for lysis (with homologous complement) of cells expressing high levels of the CAMPATH antigen, it was very efficient at lysing low antigen expressing CCRF-CEM cells, where the parental form (CAMPATH-1H) was non-lytic. It is possible that they may also have enhanced capacity for ADCC. Insufficient information exists yet on potency in ADCC, or performance in vivo.
Antibodies to the CD3 epsilon signalling molecule of the T-cell receptor complex have proven powerful immunosuppressants. OKT3 is widely used for the reversal of allograft rejection episodes. CD3 mAbs like OKT3 could be used more widely were it not for immunogenicity and the toxicity arising from massive cytokine release on first dose administration. We have taken our own rat anti-human CD3 antibody (YTH 12.5) and have engineered it to try to address some of the specific (eg. cytokine release) and more general (eg. antigenic modulation without cell destruction) issues that affect therapeutic antibodies. We have produced a humanised version with affinity comparable to the parent (11), and have sought two mutually exclusive directions for antibody improvement. The first was to derive non-lytic, blocking, and non-activating forms that would have efficacy without the side effects of OKT3. The second was to produce more lytic forms that might be better able to kill T-cells thus limiting the extent of cytokine release.
In order to find a form that might block TCR signalling but avoid cytokine release, we examined the mitogenicity of a series of isotype-matched variants (12). To our surprise all forms (IgGs, IgA and IgE) were mitogenic although IgG2 was less effective. To reduce FcR binding in a hIgG1 form, a mutation was introduced into Asn 297 to convert it to Ala; this destroys the carbohydrate attachment site and results in the synthesis of an aglycosylated IgG1 heavy chain. This mutant was non-mitogenic in vitro, yet powerful at inhibiting T-cell stimulation by antigen.
Rapid antigenic modulation by antibody is a known contributing factor limiting the lysis of cells by antibody and complement. In the past we obtained univalent forms of antibodies from somatic hybrids secreting two light chains, and had shown (13,14) that such antibodies were better able to activate complement in vitro, were still immuno-suppressive in vivo, and yet surprisingly did not cause cytokine release in the small series studied (10 patients) (14,15 ). Given these encouraging preliminary results, we sought to create univalent antibodies by a genetic engineering route.
We derived a truncated H-chain (lacking the VH and CH1 domains) as the second antibody arm, and transfected this into the humanised parental transfectoma (11). While the parental form could not activate complement, the purified univalent antibody was found to be competent. Further analysis of univalent forms of other isotypes revealed that a univalent IgG3 form was even more lytic.
This analysis demonstrates that univalent antibodies with improved complement lytic potency can be generated through relatively simple protein engineering procedures.
(1) Reichmann L; Clark M; Waldmann H and Winter G. (1988) Reshaping human antibodies for therapy. Nature 332: 323-327
(2) Gorman, S.D., Clark, M.R., Routledge, E.G., Cobbold, S.P. & Waldmann, H. (1991). Reshaping a therapeutic CD4 antibody. Proc. Natl. Acad. Sci. 88, 4181-4185.
(3) Sims, M.J., Hassal, D.G., Brett, S., Rowan, W., Lockyer, M.J., Angel, A., Lewis, A.P., Hale, G., Waldmann, H. & Crowe, J.S. (1993). A Humanized CD18 Antibody Can Block Function Without Cell Destruction. The Journal of Immunology 51, 2296-2308.
(4) Routledge, E.G., Gorman, S.D. & Clark, M. (1993). Reshaping antibodies for therapy. In Protein Engineering of Antibody Molecules for Prophylactic and Therapeutic Applications in Man (Edited by Mike Clark, Cambridge). Academic Titles
(5) Bruggemann, M; Williams GT; Bindon CI; et al. (1987) Comparison of the effector function of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. med. 1987: 166: 1351-1361
(6) Greenwood, J., Clark, M. & Waldmann, H. (1993). Structural motifs involved in human IgG antibody effector functions. Eur. J. of Immunol, 23, 1098-1104
(7) Greenwood, J. & Clark, M. (1993). Effector functions of matched sets of recombinant human IgG subclass antibodies. In: Protein Engineering of Antibody Molecules for Prophylactic and Therapeutic Applications in Man” (Edited by Mike Clark, Cambridge). pp. 85-100. Academic Titles.
(8) Morrison SL; Caufield SM and Mi-Hua Tao (1993) Complement activation and Fc receptor binding by IgG. in: Protein Engineering of Antibody Molecules fro Prophylactic and Therapeutic Applications in Man. Ed M. Clarkp 101-115. Academic Titles.
(9) Greenwood, J., Gorman, S. D., Routledge, E.G., Lloyd, I.S. & Waldmann , H. (1995). Engineering multiple domain forms of the therapeutic antibody CAMPATH-1H : Effects on complement lysis (In preparation).
(10) Shopes-B (1992) A genetically engineered human IgG mutant with enhanced cytolytic activity. J-Immunol. 1992 1; 148(9): 2918-22
(11) Routledge, E.G., Lloyd, I., Gorman, S., Clark, M. & Waldmann, H. (1991). A Humanized Monovalent CD3 Antibody which can Activate Homologous Complement. Eur. J. Immunol. 21, 2717-2725.
(12) Bolt, S., Routledge, E., Lloyd, I., Chatenoud, L., Pope, H., Gorman, S., Clark, M. & Waldmann, H. (1993). The generation of humanized, non-mitogenic CD3 monoclonal antibody which retains in-vitro immunosuppressive properties. Europ. J. Immunol. 23, 403-411.
(13) Cobbold, S.P. & Waldmann, H. (1984). Therapeutic potential of monovalent monoclonal antibodies. Nature 308, 460-462.
(14) Clark, M., Bindon, C., Dyer, M., Friend, P., Hale, G., Cobbold, S., Calne, R. & Waldmann, H. (1989). The improved lytic function and in vivo efficacy of monovalent monoclonal CD3 antibodies. Eur. J. Immunol. 19, 381-388.
(15) Abbs IC; Clark M; Waldmann H; Chatenoud L; Williams DG; Ogg CS;Koffman GC; Cameron JS and Sacks SH. (1995) Sparing of the first dose effect following monovalent anti-CD3 antibody use in allograft rejection in association with diminished release of proinflammatory cytokines. (submitted).
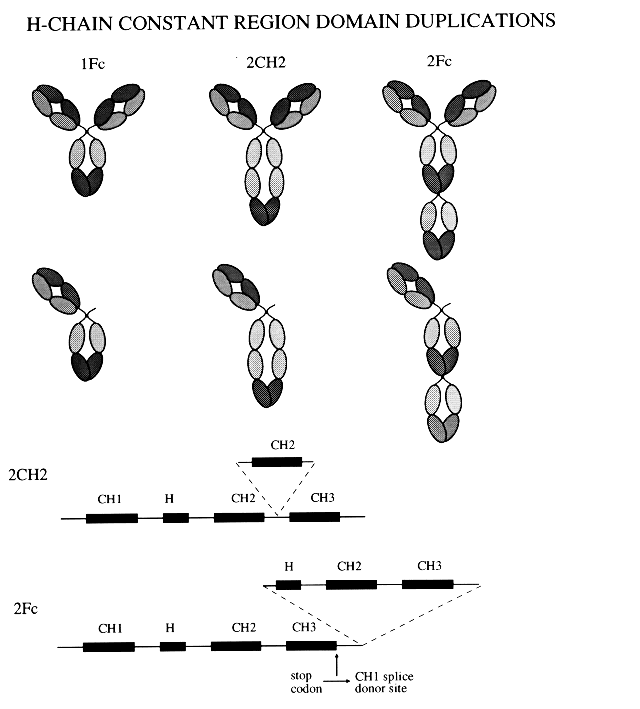 Figure 2.
Figure 2.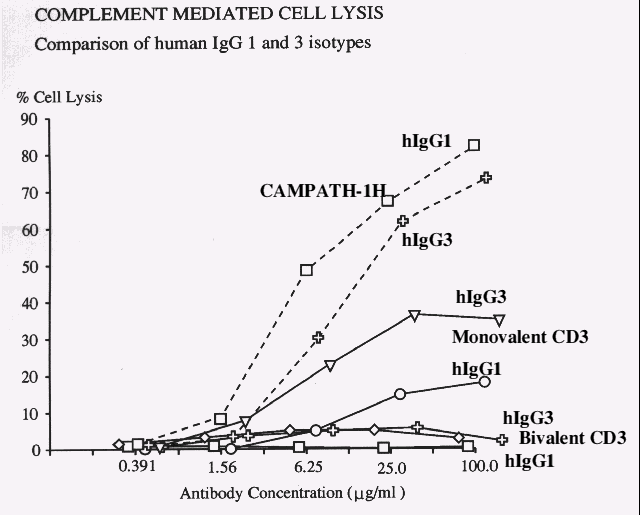 Figure 3.
Figure 3.