Research topics:
Precisely controlled gene expression is a fundamental process for the survival of any organism. It is achieved through tight interconnection between transcription, RNA processing and translation, ensuring both efficiency and fidelity of gene expression. Now it has become clear that RNA molecules are key regulatory elements in this process, raising the importance of the RNA processing step in gene expression pathway. Importantly, mutations in components regulating RNA processing can lead to global mis-regulation of gene expression and cause human diseases. Research in our lab focuses on understanding the mechanisms governing gene regulation in humans in health and disease conditions.

-
1)Molecular mechanisms underlying the pathology of neurodegeneration in humans.
-
• Motor neurone diseases
Ataxia oculomotor apraxia type 2 (AOA2) and amyotrophic lateral sclerosis type 4 (ALS4), are highly disabling disorders, representing the second most frequent type of recessive ataxias, after Friedreich’s ataxia. AOA2/ALS4 are characterised by neurodegeneration in the brain and spinal cord, causing progressive muscle weakness and finally atrophy. The gene mutated in these diseases encodes a protein called senataxin. Since the discovery of mutations causing AOA2/ALS4 diseases in 2004, little progress has been made to characterize the function of senataxin. Our recent work suggested that senataxin is involved in transcriptional regulation of gene expression (Skourti-Stathaki et al., 2011). This project will combine specialised RNA molecular approaches with the cutting edge high throughput technology to further characterize the molecular function of senataxin in humans in normal and disease conditions. In particular, we will perform experiments in cells which have been depleted from senataxin protein and neuronal cell lines established from the patients. The long term goal of this project is to develop AOA2/ALS4 molecular therapies, which will improve health and quality of patients’ life.
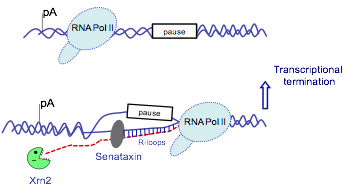
Model for the role of senataxin in transcriptional termination (from Skourti-Stathaki et al, 2011).
-
• Trinucleotide expansion diseases: Friedreich’s ataxia and Fragile X syndrome
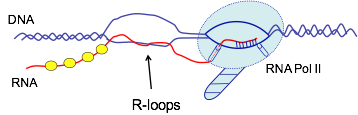
Around forty human diseases are associated with expanded repeat sequences. Among them are Friedreich ataxia and Fragile X syndrome. Friedreich ataxia (FRDA) is the most common inherited ataxia, characterised by progressive sensory ataxia, cardiomyopathy, diabetes, and premature death. FRDA is caused by an expanded GAA triplet repeat sequence in frataxin (FXN) gene, resulting in a transcriptional defect and consequently frataxin protein deficiency. Fragile X syndrome (FXS) is associated with expansion of more than 200 CGG repeats in the FMR1 gene, resulting in transcriptional silencing of FMR1 gene. This leads to dramatic reduction of FMRP protein and causes intellectual disability in patients. The molecular mechanism of FXN and FMR1 transcriptional silencing is not well understood. It has been proposed that formation of repressive heterochromatin and unusual DNA structures, such as RNA/DNA hybrids (R-loops) and triplex DNA, plays a major role in this process. This project will employ various molecular and cell biology approaches to study repeat-mediated transcriptional silencing of FXN and FMR1genes. The results generated in this project will help us to understand the molecular pathology of FRDA, FXS and other trinucleotide expansion disorders. In the long term the findings from this project will be essential for the development of new therapeutic approaches for Friedreich’s ataxia and Fragile X disorders.
2) Role of RNA/DNA hybrids (R-loops) in the regulation of human gene expression.
RNA/DNA hybrids (or R-loops) are molecular structures which are formed during the process of transcription between the nascent RNA transcript and single stranded DNA template behind the elongating RNA polymerase II. These structures are detected in E.coli, yeast and mammalian cells and they have been implicated in the processes of DNA repair, replication, transcription and recombination. Recently we demonstrated that senataxin, an RNA/DNA helicase, mutated in human neurodegenerative ataxias, can unwind R-loops, involved in the process of transcriptional termination (Skourti-Stathaki et al., 2011). This project will investigate the role of R-loops in gene expression, and uncover molecular factors and mechanisms involved in their formation in humans, employing gene-specific and whole-genome approaches.
References:
Motor neurone disease
-
1.Palau, F. and C. Espinos, Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis, 2006. 1: p. 47.
-
2.Moreira, M.C., et al., Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet, 2004. 36(3): p. 225-7.
-
3.Fogel, B.L. and S. Perlman, Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology, 2006. 67(11): p. 2083-4.
-
4.Anheim, M., et al (2009). Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 132, 2688-2698.
-
5.Criscuolo, C., et al., Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology, 2006. 66(8): p. 1207-10.
-
6.Chen, Y.Z., et al., Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol Dis, 2006. 23(1): p. 97-108.
Friedreich’s ataxia and Fragile X syndrome
-
7.Groh, M., Lufino, MM., Wade-Martins, M., Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet, (2014) May 1;10(5):e1004318.
-
8.Kumari, D., and Usdin, K. (2012). Is Friedreich ataxia an epigenetic disorder? Clin Epigenetics 4, 2.
-
9.Santoro, M.R., Bray, S.M., and Warren, S.T. (2011). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol 7, 219-245.
Sen1p, yeast homologue of senataxin
-
10.Rasmussen, T.P. and M.R. Culbertson, The putative nucleic acid helicase Sen1p is required for formation and stability of termini and for maximal rates of synthesis and levels of accumulation of small nucleolar RNAs in Saccharomyces cerevisiae. Mol Cell Biol, 1998. 18(12): p. 6885-96.
-
11.Steinmetz, E.J., et al., Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell, 2006. 24(5): p. 735-46.
-
12.Ursic, D., et al., Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res, 2004. 32(8): p. 2441-52.
-
13.Kawauchi, J., et al., Budding yeast RNA polymerases I and II employ parallel mechanisms of transcriptional termination. Genes Dev, 2008. 22(8): p. 1082-92.
Senataxin function
-
14.Suraweera, A., et al., Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol, 2007. 177(6): p. 969-79.
-
15.Suraweera, A., et al., Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet, 2009. 18(18): p. 3384-96.
-
16.K Skourti-Stathaki, N. J. Proudfoot and N. Gromak ‘Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination’. Molecular Cell (2011) 42(6), 794-805
R-loops
-
17.Aguilera, A., and Garcia-Muse, T. (2012). R loops: from transcription byproducts to threats to genome stability. Mol Cell 46, 115-124.
-
18.M.Groh, N.Gromak. Out of balance: R-loops in human disease. PLoS Genet. (2014) Sep 18;10(9).
-
19.M.Groh, L.M.Silva and N.Gromak. Mechanisms of transcriptional dysregulation in repeat expansion disorders. Biochemical Society Transactions (2014). 42(4):1123-8.
-
20.Yu, K., et al., Detection and structural analysis of R-loops. Methods Enzymol, 2006. 409: p. 316-29.
-
21.Boguslawski, S.J., et al., Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods, 1986. 89(1): p. 123-30.
-
22.El Hage, A., et al., Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev, 2011. 24(14): p. 1546-58.
-
23.Huertas, P. and A. Aguilera, Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell, 2003. 12(3): p. 711-21.
Transcriptional termination
-
24.N. Gromak., M. Dye, and N.J. Prodfoot, Pause sites promote transcriptional termination of mammalian RNA Polymerase II. Mol Cell Biol. (2006) May;26(10):3986-96
-
25.West, S., N. Gromak, and N.J. Proudfoot, Human 5' --> 3' exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature, 2004. 432(7016): p. 522-5.
-
26.Richard, P. and J.L. Manley, Transcription termination by nuclear RNA polymerases. Genes Dev, 2009. 23(11): p. 1247-69.